
La enfermedad de Pompe es un trastorno poco común, hereditario genéticamente, que afecta a alrededor de 1 de cada 40.000 personas en los Estados UnidosEsta condición afecta la capacidad de los músculos para funcionar normalmente, provocando que se debiliten con el tiempo.
La enfermedad de Pompe puede ser mortal si se diagnostica a una edad muy temprana. Aunque aún no existe cura, ciertos tratamientos y cuidados paliativos pueden ayudar a controlar los síntomas y mejorar la calidad de vida de los pacientes.
Este artículo analiza todo lo que debe saber sobre esta enfermedad hereditaria, como qué es, por qué se produce, los diferentes tipos y síntomas, y cómo se diagnostica y se trata.
¿Qué es la enfermedad de Pompe?
La enfermedad de Pompe, también conocida como enfermedad de almacenamiento de glucógeno tipo II (GSD2), es una condición genética que ocurre cuando el cuerpo no puede descomponer el glucógeno (un azúcar complejo) debido a la deficiencia de una enzima digestiva llamada alfa-glucosidasa ácida (GAA).
La enfermedad de Pompe se considera una enfermedad progresiva, lo que significa que los síntomas pueden empeorar con el tiempo si no se trata a tiempo.
Esta condición también se conoce por otros nombres, como deficiencia de alfa-glucosidasa ácida (GAA) o deficiencia de maltasa ácida (maltasa ácida es otro nombre para la alfa-glucosidasa ácida).
¿Qué causa la enfermedad de Pompe?
La enfermedad de Pompe es causada por una mutación en el gen GAA, responsable de la producción de la enzima alfa-glucosidasa ácida (GAA). El cuerpo utiliza esta enzima para descomponer el azúcar complejo (glucógeno).
Normalmente, la enzima GAA está presente en lisosomas (Pequeñas estructuras que funcionan como centros de reciclaje dentro de las células). Dentro de los lisosomas celulares, la enzima GAA ayuda a descomponer el glucógeno en glucosa, la principal fuente de energía para la mayoría de las células del cuerpo.
Sin embargo, en personas con enfermedad de Pompe, una deficiencia o baja producción de la enzima GAA debido a una mutación en el gen GAA provoca la acumulación de glucógeno en los lisosomas. Esta acumulación daña los órganos y tejidos de todo el cuerpo, en particular los músculos, lo que provoca los signos y síntomas progresivos de la enfermedad de Pompe.
Tipos de enfermedad de Pompe
La enfermedad de Pompe puede afectar a personas de cualquier edad, desde recién nacidos hasta adultos. Este trastorno se clasifica en dos tipos según la cantidad de enzima producida y el momento en que aparecen los primeros síntomas:
1. Enfermedad de Pompe de inicio infantil (EPI)
La enfermedad de Pompe de inicio infantil o temprana es más grave y progresa con mayor rapidez que la de inicio tardío. En pacientes con este tipo, la enzima alfa-glucosidasa ácida se produce en cantidades muy bajas o no se produce en absoluto.
La enfermedad de inicio infantil se divide a su vez en dos subtipos según el momento en que aparecen los primeros síntomas. Por ejemplo:
Inicio infantil clásico
En este tipo, los síntomas comienzan a aparecer durante los primeros meses de vida, con mayor frecuencia entre los 2 y los 4 meses de edad. A los 4 años, se produce daño muscular en el corazón, lo que provoca cardiomegalia (agrandamiento del corazón) y, finalmente, insuficiencia cardíaca.
Además, también pueden presentarse debilidad muscular progresiva (miopatía), tono muscular deficiente (hipotonía), hepatomegalia (aumento del tamaño del hígado) y cardiopatías. Los bebés afectados tampoco logran aumentar de peso ni crecer con normalidad.
Inicio infantil no clásico
Los bebés tienden a mostrar signos de esta enfermedad rara al cumplir un año. Experimentan debilidad muscular progresiva y retraso en las habilidades motoras. Esta debilidad muscular también causa problemas respiratorios en los bebés con el tipo no clásico.
Sin tratamiento, puede ser potencialmente mortal durante el primer año debido a problemas respiratorios.
2. Enfermedad de Pompe de inicio tardío (LOPD)
La enfermedad de Pompe juvenil o adulta (LOPD), también conocida como enfermedad de Pompe, puede presentarse a cualquier edad. Este tipo es menos grave que la de inicio infantil. Aunque progresa lentamente, puede causar discapacidad grave.
Las personas con el tipo LOPD producen una cantidad baja de enzima alfa-glucosidasa ácida (GAA) y los primeros síntomas pueden aparecer típicamente más tarde en la niñez, la adolescencia o la adultez.
Los pacientes con enfermedad de Pompe de inicio infantil suelen experimentar debilidad muscular progresiva (miopatía) que puede provocar problemas respiratorios y, eventualmente, insuficiencia respiratoria. A diferencia de la enfermedad de Pompe de inicio infantil, este tipo tiene menos probabilidad de afectar el corazón.
¿Cuáles son los signos y síntomas comunes?
Como se mencionó anteriormente, los síntomas de esta enfermedad pueden variar según el tipo y la etapa de la misma. Algunos síntomas comunes incluyen:
- Debilidad muscular, especialmente en el tronco, hombros, caderas y piernas.
- Tono muscular deficiente (hipotonía), donde los músculos se sienten flácidos.
- Dificultad para respirar debido al debilitamiento del diafragma y de los músculos respiratorios.
- Agrandamiento del corazón (más común en la forma infantil).
- Problemas de alimentación como dificultad para succionar, tragar o masticar.
- Retraso en las habilidades motoras, como darse la vuelta, sentarse o caminar.
- Fatiga, como cansancio constante y baja resistencia.
- Escoliosis (curvatura de la columna vertebral debido al debilitamiento de los músculos).
¿Cómo se diagnostica la enfermedad de Pompe?
El diagnóstico temprano y preciso es fundamental, especialmente si los síntomas de este trastorno aparecen unos meses después del nacimiento. Las pruebas más definitivas y específicas para la enfermedad de Pompe incluyen la biopsia muscular, el análisis de sangre y el análisis genético molecular.
1. Biopsia muscular
En la enfermedad de Pompe, una biopsia muscular a menudo muestra fibras musculares con pequeñas bolsas llenas de exceso de glucógeno.
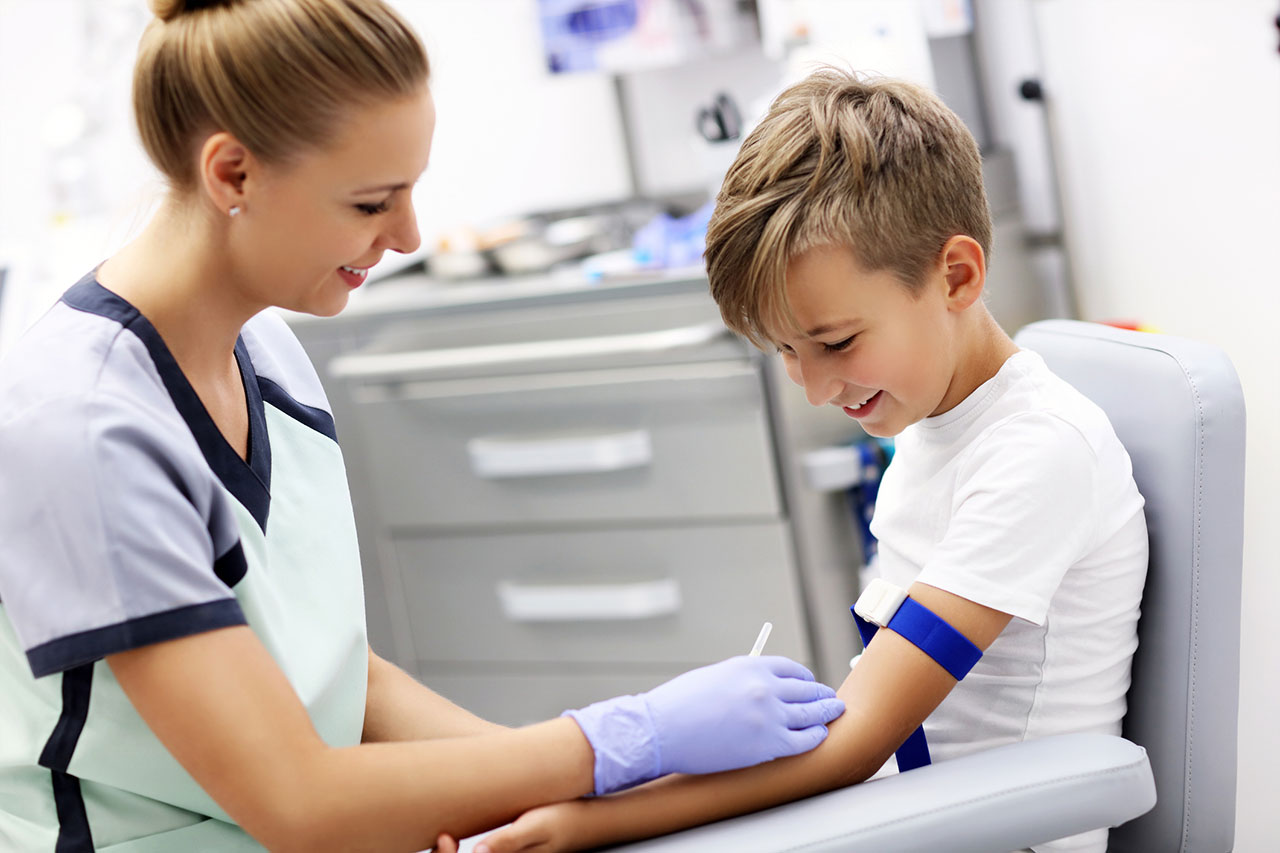
2. Análisis de sangre
Un análisis de sangre mide la actividad de la enzima α-glucosidasa ácida (GAA) mediante una muestra de sangre o una biopsia de piel o músculo. Los niveles bajos de la enzima GAA en la sangre del paciente pueden ayudar a diagnosticar la enfermedad de Pompe.
3. Pruebas genéticas moleculares
Las pruebas genéticas moleculares son el segundo paso para confirmar el diagnóstico de la enfermedad por almacenamiento de glucógeno tipo II (GSDII). Esto ayuda a identificar las mutaciones en el gen GAA. Actualmente, se han identificado entre 300 y 500 mutaciones de GAA, y las mutaciones específicas varían entre la IOP y la LOOP.
Además de los análisis de sangre y el análisis genético, es posible que también le realicen otras pruebas de laboratorio para detectar o evaluar los síntomas potencialmente asociados con esta enfermedad. Esto incluye:
- Creatina quinasa sérica (CK): Para comprobar el nivel de la enzima creatina quinasa en la sangre.
- Electromiografía (EMG)Esta prueba mide la actividad eléctrica muscular. En otras palabras, evalúa el funcionamiento de los músculos.
- resonancias magnéticas o tomografías computarizadas:Para comprobar si hay daño muscular.
- Ecocardiograma (Eco) y radiografías de tórax: A evaluar la función cardíaca y el tamaño del corazón, que está agrandado en la enfermedad de Pompe infantil clásica.
- Pruebas de función pulmonarPruebas respiratorias que miden la capacidad pulmonar. Si padece la enfermedad de Pompe en adultos o jóvenes, puede solicitar esta prueba, ya que la insuficiencia respiratoria es frecuente.
¿Cuáles son las opciones de tratamiento disponibles?
Aunque no existe cura para la enfermedad de Pompe, generalmente se recomiendan tratamientos estándar como la terapia de reemplazo enzimático (TRE) para retardar la progresión de la enfermedad, mejorar los síntomas y prolongar la expectativa de vida de los pacientes.
1. Terapia de reemplazo enzimático (TRE)
La terapia de reemplazo hormonal (ERT) es un tratamiento estándar para todos los pacientes con diagnóstico de enfermedad de Pompe. En esta terapia, la alfa-glucosidasa ácida humana recombinante (rhGAA), como Alglucosidasa alfa (indicado para IOPD y LOPD) y Avalglucosidasa alfa (indicado para la enfermedad de Parkinson), se administra por vía intravenosa a los pacientes. Estos fármacos Son enzimas modificadas genéticamente que imitan la función de la enzima GAA normal y ayudan a:
- Reduce la acumulación de glucógeno descomponiéndolo en azúcar simple (glucosa).
- Disminuir el tamaño del corazón y mantener su función normal.
- Mejorar la función muscular, el tono y la fuerza o al menos retrasar la progresión del trastorno.
Además, en 2023, la FDA aprobó la terapia combinada de cipaglucosidasa alfa-atga (Pombiliti) y miglustat (Opfolda) para tratar a adultos con enfermedad de Pompe de aparición tardía que pesan ≥40 kg y tienen síntomas que no mejoran con su terapia de reemplazo enzimático actual.
2. Tratamientos adicionales
Además del tratamiento estándar, también es posible que un equipo de tratamiento recomiende algunas terapias de apoyo, como:
- Terapia respiratoria (si los pacientes tienen algún grado de insuficiencia respiratoria)
- Fisioterapia (puede utilizarse para fortalecer los músculos respiratorios)
- Terapia ocupacional (para ayudar con las actividades diarias)
- Terapia nutricional (incluyendo sondas de alimentación para bebés o ajustes dietéticos para adultos si se ven afectados los músculos de la masticación y la deglución)
¿Cuál es la esperanza de vida de una persona con enfermedad de Pompe?
El pronóstico para las personas con esta enfermedad depende del tipo y de la prontitud con la que se inicie el tratamiento. Por ejemplo, los bebés con inicio infantil suelen sobrevivir menos de dos años debido a insuficiencia respiratoria o cardíaca si no se diagnostican y tratan a tiempo.
Con ERT, muchos niños vivir más tiempo y tener una función cardíaca mejorada, aunque la debilidad muscular aún puede progresar.
Por otro lado, la progresión de la enfermedad de Pompe en la edad adulta es más lenta y puede producirse insuficiencia respiratoria si los músculos respiratorios se debilitan significativamente.
¿Podemos prevenir la enfermedad de Pompe?
No, no se puede prevenir la enfermedad de Pompe porque es una condición genética que se transmite de padres a hijos con un patrón autosómico recesivo, lo que significa que usted puede contraer esta enfermedad si recibe dos copias de la GAA mutada de cada uno de sus padres portadores.
El asesoramiento genético, la detección de portadores y las pruebas prenatales o preimplantacionales pueden ayudarle a comprender su riesgo de tener un hijo con la enfermedad de Pompe y respaldar la toma de decisiones informada.
REFERENCIAS:
- Stevens, D., Milani-Nejad, S., y Mozaffar, T. (2022). Enfermedad de Pompe: Panorama clínico, diagnóstico y terapéutico. Opciones de tratamiento actuales en neurología, 24(11), 573. https://doi.org/10.1007/s11940-022-00736-1
- Chien, Y., Hwu, W. y Lee, N. (2013). Enfermedad de Pompe: el diagnóstico y el tratamiento tempranos marcan la diferencia. Pediatría y Neonatología, 54(4), 219-227. https://doi.org/10.1016/j.pedneo.2013.03.009
- Kohler, L., Puertollano, R., y Raben, N. (2018). Enfermedad de Pompe: de la ciencia básica a la terapia. Neuroterapéutica, 15(4), 928. https://doi.org/10.1007/s13311-018-0655-y
- Morales, A., De Melo, MHST, Anastasopoulou, C. y Anilkumar, AC (28 de enero de 2025). Enfermedad de almacenamiento de glucógeno tipo II. StatPearls - Estantería NCBI. https://www.ncbi.nlm.nih.gov/books/NBK470558/
- Leslie, N., y Bailey, L. (2 de noviembre de 2023). Enfermedad de PompeGeneReviews® – Biblioteca del NCBI. https://www.ncbi.nlm.nih.gov/books/NBK1261/
- Meena, NK, y Raben, N. (2020). Enfermedad de Pompe: nuevos avances en un antiguo trastorno de almacenamiento lisosomal. Biomoléculas, 10(9), 1339. https://doi.org/10.3390/biom10091339
- Lim, J., Li, L. y Raben, N. (2014). Enfermedad de Pompe: de la fisiopatología a la terapia y viceversa. Fronteras en la neurociencia del envejecimiento, 6, 103952. https://doi.org/10.3389/fnagi.2014.00177
- Labella, B., Cotti Piccinelli, S., Risi, B., Caria, F., Damioli, S., Bertella, E., Poli, L., Padovani, A. y Filosto, M. (2023). Una actualización completa sobre la enfermedad de Pompe de aparición tardía. Biomoléculas, 13(9), 1279. https://doi.org/10.3390/biom13091279

